Hello
I have a dataset with 1800 samples and 600m variants. I need to do phewas for 1300 phenotypes. I am using variants with MAF > 5%(few million variants) and i am running as below with one phenotype at a time. I tried it for first 50 phenotypes and got output for 15 phenotypes which took me 7 hours (20masters/100preempts) is there a way to speedup the process to make it cost effective? and also can you review the code.(already kept in zulip keeping here too) Thank you.
Code:
#!/usr/bin/env python
# coding: utf-8
import hail as hl
import hail.expr.aggregators as agg
hl.init(default_reference = "GRCh38")
import numpy as np
import pandas as pd
from collections import Counter
from math import log, isnan
from pprint import pprint
from bokeh.io import show, output_notebook
from bokeh.layouts import gridplot
output_notebook()
#Code to generate a smaller JHS fileset
mt = hl.read_matrix_table('gs://jhs_data_topmed/topmed_6a_pass_2k_minDP10_sQC_vQC_AF01_jhsprot.mt')
###variants to keep after ld prune
var = hl.import_table('gs://jhs_data_topmed/variants.tsv', impute=True,key="v")
var_to_keep = var.key_by(**hl.parse_variant(var.v))
prune = mt.filter_rows(hl.is_defined(var_to_keep[mt.row_key]) , keep=True)
print(prune.count())
###perform rrm once
rrm = hl.realized_relationship_matrix(prune.GT).to_numpy()
####lmm for multiple phenotypes(linear tooo)
list = [prune.pheno.SL003310,prune.pheno.SL004140,prune.pheno.SL004146,prune.pheno.SL004258,prune.pheno.SL004557,prune.pheno.SL004725,prune.pheno.SL004726,prune.pheno.SL004858,prune.pheno.SL004939]
list1 = ['SL003310','SL004140','SL004146','SL004258','SL004557','SL004725','SL004726','SL004858','SL004939','SL005152','SL005153','SL005208','SL005213','SL005256','SL005430','SL005574','SL005699']
### multiple phenotypes linear regression mixed model
for i in range(0,46):
name = list[i]
print(name)
out = list1[i]
print(out)
model,p = hl.linear_mixed_model(y=name,x=[1.0,prune.pheno.male,prune.pheno.age],k=rrm,p_path='gs://jhs_data_topmed/lmm_phewas/'+out+'.bm')
model.fit()
model.h_sq
result_table = hl.linear_mixed_regression_rows(mt.GT.n_alt_alleles(), model)
result_table.export('gs://jhs_data_topmed/lmm_phewas/gwas.'+out+'.tsv.gz')
It looks like you’re doing the right thing by computing the kinship once and reusing it across all phenotypes.
Are all these phenotypes on exactly the same samples? If so, after you’ve run LMM once with pa_t_path set as well, use the approach documented in linear_mixed_regression_rows under the heading Efficiently varying the response or set of fixed effects in the full-rank case to run it all subsequent times. This will avoid recomputing pa_t each time, which is the most intensive step by far, so you should see a huge speed-up on all subsequent runs.
Other questions once you’ve made this change:
For each loop:
How long does the fit step take? (should be very quick)
How much time is spent on linear_mixed_regression_rows step?
Do the loops each take the same amount of time or so they get substantially slower with time?
600 million is a LOT of variants. I’m confused how you could have that many after filtering at 5% minor allele frequency. Can you explain?
And can you test how long it takes to run a single (unmixed) linear_regression_rows for comparison so we know where the bottleneck is?
—
If it’s still slow after the major fix above, a smaller speed up would come from us upgrading our infrastructure to accommodate testing multiple phenotypes with LMM in one pass, similar to what we do for linear regression on UKBB. Knowing these numbers will help us evaluate how helpful that would be.
Aside: With so few samples, I suspect you are using more variants to estimate kinship than necessary. 50k should be plenty. If you’re using millions, you could downsample to get there. But if you already have a kinship matrix you’re happy with, this won’t speed you up since all the time is in the loop.
Are all these phenotypes on exactly the same samples? Yes,all the phenotypes have same samples and i will try the method you suggested above with fewer phenotypes.
Other questions once you’ve made this change:
For each loop:
How long does the fit step take? (should be very quick) i am thinking it was quick as i dint checked the time before
How much time is spent on linear_mixed_regression_rows step? I think it took more time but will check after changing according to your suggestion.
I will respond back once i check these.
Do the loops each take the same amount of time or so they get substantially slower with time? this is also one thing i will have a look into.
600 million is a LOT of variants(its for initial dataset). I’m confused how you could have that many after filtering at 5% minor allele frequency. Can you explain? Subsetted version have less variants compared to initial set
And can you test how long it takes to run a single (unmixed)
linear_regression_rows for comparison so we know where the bottleneck is? ok will have a look into it and check too
Aside: With so few samples, I suspect you are using more variants to estimate kinship than necessary. 50k should be plenty. If you’re using millions, you could downsample to get there. But if you already have a kinship matrix you’re happy with, this won’t speed you up since all the time is in the loop.
Hi Akhil, when you run linear_mixed_regression_rows the first time, you need to set the pa_t_path to store this block matrix once and for all. Then to GWAS other phenotypes, you want to pass in pa_t_path, not p_path, to fit_alternatives as in the documentation.
This may be what’s causing the error message far above, which is not about missing values; it’s a dimension mismatch for matrix multiplication. If you’re still getting this error, check the dimensions of p, y, and x in your case with .shape
Can you post your new code and tell me what shapes you’re getting?
You should be running linear_mixed_regression_rows once, saving out pa_t_path. You can then do the other phenotypes in a loop with LinearMixedModel and fit_alternatives. But before you do the rest in a loop, make sure that the latter works on the same phenotype, giving the same results as linear_mixed_regression_rows but much faster because it doesn’t recompute the product of p and a_t.
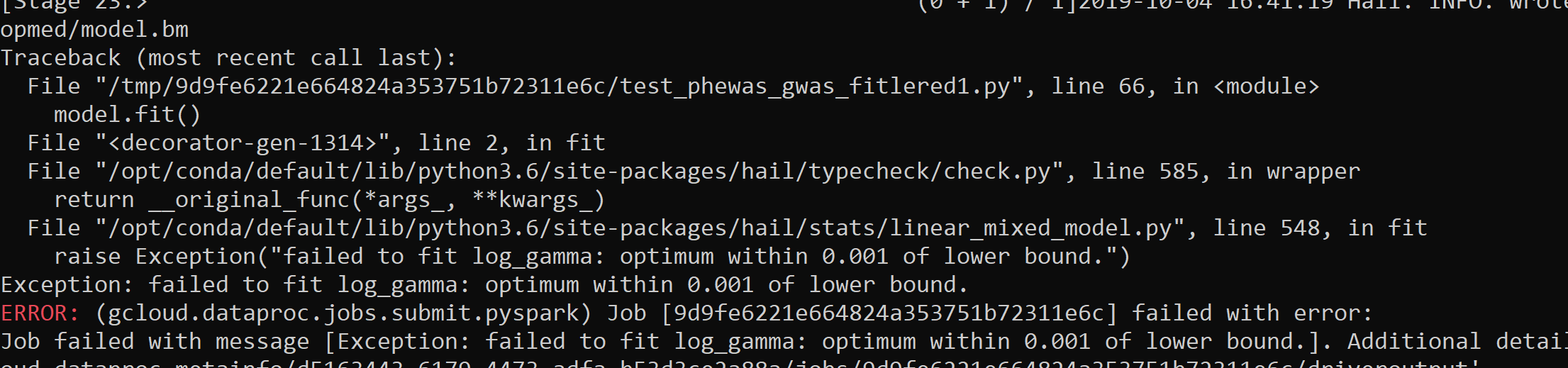
The picture of the google cloud screen you posted is cutoff in a way that makes it impossible to read the output. Posting text is also generally better than posting pictures.