Hi,
I’m trying to open a gvcf file as a matrix table, but I’m not sure that I’m doing it in the right way.
I’m defining the partitions by the regions list:
CHR_RANGES = {'chr1': 248956422, 'chr2': 242193529, 'chr3': 198295559, 'chr4': 190214555, 'chr5': 181538259,
'chr6': 170805979, 'chr7': 159345973, 'chr8': 145138636, 'chr9': 138394717, 'chr10': 133797422,
'chr11': 135086622, 'chr12': 133275309, 'chr13': 114364328, 'chr14': 107043718, 'chr15': 101991189,
'chr16': 90338345, 'chr17': 83257441, 'chr18': 80373285, 'chr19': 58617616, 'chr20': 64444167,
'chr21': 46709983 , 'chr22': 50818468, 'chrX': 156040895, 'chrY': 57227415}
regions = [hl.parse_locus_interval("[chr1:1-chr1:248956422]")]
for chrom in CHR_RANGES.keys():
if chrom != 'chr1':
interval = hl.parse_locus_interval("[" + chrom + ":1-" + chrom + ":" + str(CHR_RANGES[chrom]) + "]")
regions.append(interval)
Then:
PKGen160 = hl.import_gvcfs(['gvcf_path'], partitions=regions)
And these are the first 10 rows of the matrix table that I got:
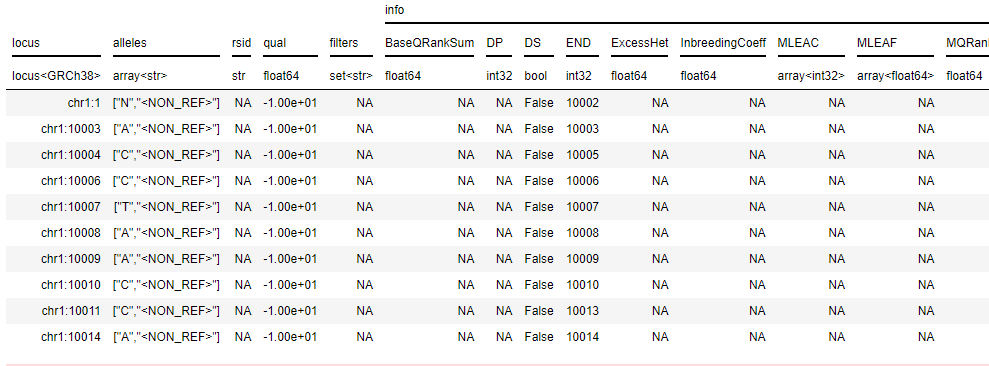
Is it ok?
Thanks,
Ofer